Configuring and customizing a SequenceServer BLAST installation
Install on Mac/Linux/Unix or use our Cloud service
Requirements for local installation
- Linux or Mac and Ruby (3.2)
- NCBI BLAST+ (2.14.0+) is interactively downloaded if absent
-
Standard Unix build
tools (e.g.,
gcc,make) are required to install SequenceServer. This is because SequenceServer's need to parse BLAST's XML output compiles some C code as part of the installation process. This means that the On a Mac, this means having Xcode and CLI tools for Xcode installed. On Ubuntu and other Debian-based Linux systems, you would have to install theruby-devandbuild-essentialpackages in addition toruby. - Alternatively, use Docker to run SequenceServer on Linux, Mac, or Windows.
- Alternatively, use our cloud BLAST service: forget about the command-line and servers; use a point-and-click interface to set up everything in a secure manner, with powerful servers accessible worldwide.
Install or update
Once you have Ruby and the build tools installed, or Docker, the commands below can be used to install SequenceServer for the first time. Later on, the same command can be used to update SequenceServer to the latest version:
sudo gem install sequenceserver If using Docker:
docker pull wurmlab/sequenceserver Configure and run
Run the following in a terminal to configure and run SequenceServer. It will automatically download NCBI BLAST+ if absent, ask for the location of directory containing database sequences, format FASTA files for use with BLAST+, and list them for use in the search form:
sequenceserver If using Docker, you need to provide the databases directory up-front:
docker run -itp 4567:4567 -v /path-to-database-dir:/db wurmlab/sequenceserver
That's it! Open http://localhost:4567 in your web-browser and start BLAST-ing!
Where is SequenceServer installed?
This varies from computer to computer. Run the following command in a terminal to find out:
echo "$(ruby -e 'puts Gem.path[0]')/gems/sequenceserver-2.1.0"
You may need to change 2.0.0 in the above command to
reflect the version of SequenceServer you are running.
Basics of configuring SequenceServer
SequenceServer requires the location of NCBI BLAST+ binaries and the
location of database sequences (either in FASTA or BLAST database
format) to run. These can be specified using command line parameters
or through a configuration file.
SequenceServer looks for a configuration file by default at
~/.sequenceserver.conf. This can be changed by
using the -c option:
sequenceserver -c ~/.sequenceserver.ants.conf.
Configuration files have a simple key-value syntax and can be viewed
and modified with standard tools. Alternatively, -s option
can be used to add an arbitrary key-value to the configuration file or
to change the value of a key:
sequenceserver -c ~/.sequenceserver.ants.conf -s -d /path/to/new/location/of/database/sequences
sequenceserver -s -b /path/to/latest/blast/binaries
The following table lists all configuration values accepted by SequenceServer through the configuration file or through command line options. Command line options take precendence over the values in configuration file.
| Configuration file | Command line | Description |
|---|---|---|
| :bin: | -b / --bin | Indicates path to the BLAST+ binaries. |
| :database_dir: | -d / --database_dir | Indicates path to the BLAST+ databases. |
| :num_threads: | -n / --num_threads | Number of threads to use for BLAST search. |
| :num_jobs: | Number of BLAST searches to run concurrently (default: 1). | |
| :job_lifetime: | How long to keep search results for (in minutes). | |
| :options: | Predefined search options for different BLAST algorithms. | |
| :frame_options: |
Access options for embedding SequenceServer in an iframe. Possible
values :deny, :sameorigin, or
'ALLOW-FROM uri'.
|
|
| :require: | -r / --require | Load extension from this file. |
| :host: | -H / --host | Host to run SequenceServer on. |
| :port: | -p / --port | Port to run SequenceServer on. |
The following table lists additional command line options that are available. We have already seen the second and the third option. We will discuss the rest in following sections.
| Command line | Description |
|---|---|
| -x / --import | Import pre-generated BLAST/DIAMOND XML output for visualisation |
| -c / --config_file | Provide path location of your custom configuration file |
| -s / --set | Set configuration value in default or given config file |
| -m / --make-blast-databases | Create, update or reformat BLAST databases |
| -l / --list-databases | List found BLAST databases |
| -i / --interactive | Run SequenceServer in interactive mode |
| -D / --devel | Run SequenceServer in development (debug) mode |
| -v / --version | Print version number of SequenceServer that will be loaded |
| -h / --help | Display this help message |
Creating BLAST databases
The BLAST search algorithms don't directly understand FASTA files.
BLAST includes the makeblastdb tool that is used to
convert FASTA files into the optimized BLASTDB format, which is
then used by the search algorithms:
makeblastdb -dbtype <prot_or_nucl> -title <human_readable_name> -in <path_to_fasta> -parse_seqids
SequenceServer's makeblastdb wrapper can recursively scan a
directory for FASTA files and prompt you to convert them into BLAST
databases. SequenceServer automatically determines whether the file
contains nucleotide or amino acid sequences so you don't have to specify
it yourself and suggests a human readable name by "cleaning" the FASTA
file name.
SequenceServer does this automatically when it does not find any BLASTDB
files in database_dir. Rest of the times you can invoke
this functionality manually, such as after adding new FASTA files to
database_dir.
sequenceserver -m
The above command reads database_dir from the default
configuration file(~/.sequenceserver.conf), but that
can be changed:
sequenceserver -m -d /path/to/directory_with_fasta_files sequenceserver -m -c /path/to/config_file_containing_database_dir
If you would like to include taxonomy id of sequences in the database,
you can do so by including .taxid_map.txt file next to the
FASTA file. For example, if your FASTA file is
/database_dir/ants.fa, the taxid map file must be called
/database_dir/ants.taxid_map.txt. If you have this file,
SequenceServer will automatically use it with
-taxid_map option of makeblastdb. The file is
expected to contain a sequence id and a taxonomy id on each line.
If you do not have this file, SequenceSever will prompt you to enter one taxonomy id that can be used for all sequences in the FASTA file. You can get the taxonomy id of a species at NCBI Taxonomy browser.
An example prompt:
FASTA file: /Users/priyam/biodb/protein/Solenopsis_invicta/SI2.2.3.fa FASTA type: protein Proceed? [y/n] (Default: y): Enter a database title or will use 'SI 2.2.3 ': Enter taxid (optional): 13686
Aroon Chande has put together a script to automatically create BLASTDBs and restart SequenceServer when a FASTA file is added to database directory.
Upgrading BLAST databases
NCBI has introduced a new BLAST database format, called version 5. If
you have a mix of the old, version 4, and version 5 databases in your
databases directory, it can cause unexpected problems. Furthermore,
for features like FASTA download to work correctly, it is important
that BLAST databases are created using the -parse_seqids
option of makeblastdb.
SequenceServer checks for such incompatibilities automatically on startup and offers to upgrade problematic databases. This works even if you have lost the original FASTA file from which the database was created. Human readable database name and taxonomy identifiers in the databases are preserved during the upgrade. Note that you may find intermediate FASTA and taxid map files in the databases directory after upgrading databases.
You can also invoke this functionality by running sequenceserver
-m.
Using BLAST databases from NCBI
NCBI provides publicly available sequences as pre-formatted BLAST
databases and can be downloaded with update_blastdb.pl
script distributed with BLAST. Since these databases are huge, they
are split across several files (volumes) and linked together with an
alias file. SequenceServer works seamlessly with such, multi-part
databases. We also have an alternative to
update_blastdb.pl to download BLAST databases from NCBI
faster: ncbi-blast-dbs.
# Install ncbi-blast-dbs
sudo gem install ncbi-blast-dbs
# View available BLAST databases.
ncbi-blast-dbs
# Download one or more databases.
ncbi-blast-dbs nt nrFurther, SequenceServer understands NCBI sequence ids and automatically links to NCBI page corresponding to the hit sequences from the HTML report.
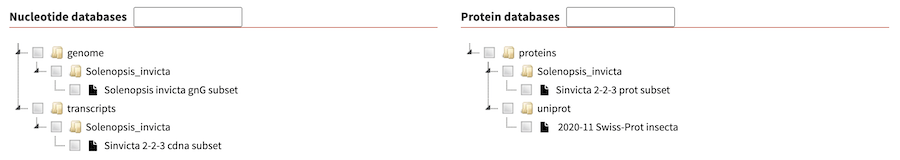
Tree widget for databases
If you have a long list of databases, you can use the experimental 'tree widget' for displaying databases that was contributed by Björn Hammesfahr of KWS SAAT SE & Co. KGaA.
To enable it, change the :database_widget: key in
configuration file to tree.
:databases_widget: treeThis folder mimics the structure of the databases directory and respects symlinks. You can find example directly structure and screenshot in the above mentioned link.
As a further example, the example database dir include in SequenceServer code base looks as follows with the tree data widget:
Advanced BLAST options
With a few exceptions, all command-line BLAST+ parameters can be
provided using the "Advanced params" textbox in the search form.
Options that change input/output behaviour (e.g.,
-query, -db, -subject,
-outfmt, -import_search_strategy) are
not allowed.
For security, only letters, numbers, space, hyphen, underscore, and period are allowed in "Advanced params" textbox.
SequenceServer changes BLAST+'s default:
-
-evalue 1e-5is added to all searches -
-task blastnis added to BLASTN searches
The above changes are applied transparently, i.e., they are added to the 'Advanced params' textbox once you have pasted your query and selected the databases and can be overriden.
The advanced parameters applied by SequenceServer are listed in the configuration file. You can change them as per your requirements.
Starting with version 2.1 of SequenceServer, it is possible to define multiple advanced parameter "presets" in the config file for each BLAST algorithm that are then automatically made available in the search form. Here's an example:
:options:
:blastn:
:default:
- "-task blastn"
- "-evalue 1e-5"
:short-seq:
- "-task blastn-short"
- "-evalue 1e-1"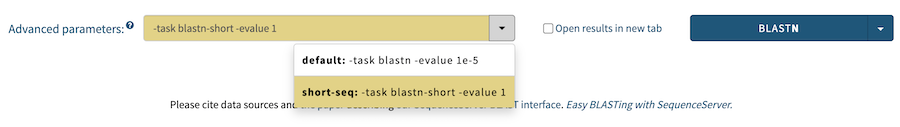
Taxonomy of matching sequences
SequenceServer automatically includes scientific name of the species in its HTML report. All taxonomy data returned by BLAST is provided in the "Full tabular report" download option. For this to work, BLAST database files should contain taxonomy id of the sequences and you must have downloaded NCBI "taxdb".
See "Creating BLAST databases" section for how to include taxonomy information in BLASTDB files. If you are using BLAST databases downloaded from NCBI then you don't need to worry about this - taxonomy information is included in the database files.
To download NCBI taxdb, run:
sequenceserver --download-taxdb
The above command downloads taxdb files to ~/.sequenceserver,
where SequenceServer keeps a few other files as well.
Adding links to search hits
It is often desirable to link search hits to external resources such as
NCBI, UniProt, or a genome browser. SequenceServer provides a powerful
and flexible mechanism to do this.
Simply edit lib/sequenceserver/links.rb in your
SequenceServer installation directory to add a link generator function,
based on examples and documentation provided in that file.
Alternatively, you can write your link generator functions in a
separate file and load it through :require_file: key in
config file.
You can access methods defined in the Hit
class within a link generator. Alignment coordinates are not defined on a hit, but on hsps. Calling
hsps method (in link generator) will return an Array of HSP objects for
that Hit.
Which database a hit came from is not provide by BLAST in it’s output. You can call out to whichdb
method from your link generator to get a list of all databases that the hit could have come from. If your
sequences have unique ids across _all_ FASTA files / BLAST databases, you know that the only element in the list
is the database that the hit came from. whichdb returns an Array of SequenceServer::Database
objects from which you can get database title and path. whichdb is slow. Alternative is to encode
db info (a short name) in the sequence id, and use regex matching to decide which database a hit came from.
URL parameters should be encoded. It replaces whitespace and other relevant chars in the string with % encoding followed in URLs.
Integrating with JBrowse
JBrowse's website has an excellent tutorial in this regard: How can I link BLAST results to JBrowse. The tutorial makes use of SequenceServer's plugin architecture which is described briefly in the previous section.
Autostart with systemd
Either put your user account or create a local user account for SequenceServer
sudo useradd -s /sbin/nologin seqservuser.
Create file /etc/systemd/system/sequenceserver.service with the following content, changing
ExecStart (and maybe User) to match your environment:
[Unit]
Description=SequenceServer server daemon
Documentation="file://sequenceserver --help" "http://sequenceserver.com/doc"
After=network.target
[Service]
Type=simple
User=seqservuser
ExecStart=/path/to/bin/sequenceserver -c /path/to/sequenceserver.conf
KillMode=process
Restart=on-failure
RestartSec=42s
RestartPreventExitStatus=255
[Install]
WantedBy=multi-user.targetStop any SequenceServer instance you might be running and check the above works by running the following command:
## let systemd know about changed files
sudo systemctl daemon-reload
## enable service for automatic start on boot
systemctl enable sequenceserver.service
## start service immediately
systemctl start sequenceserver.serviceSee systemd website for more options and debugging if it fails.
Autostart on Ubuntu / Bio Linux
Create file /etc/init/sequenceserver.conf with the
following content, changing author and
setuid lines to your name and username:
description "Upstart config for SequenceServer"
author "<full name>"
start on filesystem
stop on shutdown
setuid <username>
exec sequenceserverStop any SequenceServer instance you might be running and check the above works by running the following command:
sudo start sequenceserverSee Upstart Cookbook for more options and debugging if it fails.
Autostart on Mac OS X
Create file ~/Library/LaunchAgents/sequenceserver.plist
with the following content:
<?xml version="1.0" encoding="UTF-8"?>
<!DOCTYPE plist PUBLIC "-//Apple//DTD PLIST 1.0//EN" "http://www.apple.com/DTDs/PropertyList-1.0.dtd">
<plist version="1.0">
<dict>
<key>KeepAlive</key>
<true />
<key>Label</key>
<string>sequenceserver</string>
<key>ProgramArguments</key>
<array>
<string>/usr/local/bin/sequenceserver</string>
</array>
<key>RunAtLoad</key>
<true />
</dict>
</plist>Stop any SequenceServer instance you might be running and check the above works by running the following command:
launchctl load ~/Library/LaunchAgents/sequenceserverIntegrating with Apache
SequenceServer's built-in webserver can handle medium workloads. Though, for large communities or to integrate SequenceServer as part of existing websites it may be desirable to run SequenceServer with Apache. Also, setting up with Apache means SequenceServer will automatically be available when server restarts.
To setup SequenceServer with Apache, first install Phusion Passenger™ by following the instructions at their website.
Then configure Apache to load SequenceServer by following their guide on deploying a Ruby applicaion,
replacing /path-to-your-app with SequenceServer's installation directory. Finally, go to the
directory where SequenceServer is installed and edit config.ru to indicate absolute path to
SequenceServer's config file and DOTDIR which are respectively ~/.sequenceserver.conf
and ~/.sequenceserver by default:
# Remove this line.
SequenceServer.init
# And add these two, changing the path.
SequenceServer::DOTDIR = "/home/foo/.sequenceserver"
SequenceServer.init :config_file => "/home/foo/.sequenceserver.conf"
For SequenceServer 1.0.7 and earlier, you will additionally need to
delete Gemfile from SequenceServer's installation
directory.
If you plan to deploy multiple SequenceServer instances, you should deploy each to a sub-uri.
If you deploy to a sub-uri a trailing slash is required for JS, CSS and the icons to load properly. Ideally, just putting a trailing slash in Apache config should be sufficient. See this thread for more solutions.
Further, because BLAST searches can take time, you may additionally want to configure Timeout in your Apache config
to a suitable value (e.g., 5 minutes) so that the Apache doesn't close the connection before a BLAST search has
been performed.
Reverse proxy setup with Nginx
In reverse proxy setup, requests are forwarded from Nginx (or Apache) to SequenceServer's built-in server. Following config indicates how to proxy requests from Nginx to SequenceServer from a sub-uri of your domain (my-domain.com/sequenceserver). Nginx will timeout requests if it can't connect to SequenceServer within 8 seconds or if it doesn't hear back from SequenceServer within 180 seconds (3 minutes) after it forwarded the request (that is, BLAST requests that take more than than 3 minutes will be timed out by Nginx). Please see Nginx documentation for details info of each directive.
location /sequenceserver/ {
root /home/priyam/sequenceserver/public/dist;
proxy_pass http://localhost:4567/;
proxy_intercept_errors on;
proxy_connect_timeout 8;
proxy_read_timeout 180;
}SequenceServer can be integrated with Nginx similar to Apache, using Phusion Passenger. And Apache can be used instead of Nginx to proxy connections as well. Whether to use reverse proxy or Phusion Passenger and Apache or Nginx is up to the user. A discussion of pros and cons of each is beyond the scope of this documentation.
Password protection
If you are using SequenceServer with Apache or Nginx then you can easily password protect your data using HTTP basic authentication scheme. These tutorials from DigitalOcean detail th e steps required for both Apache and Nginx.
If you are using SequenceServer without Apache or Nginx, you can still
add password protection quite easily. Just add the following snippet at
line number 57 in lib/sequenceserver/routes.rb, change the
password ('admin') to something more and secure, and restart SequenceServer.
use Rack::Auth::Basic, "Restricted Area" do |username, password|
username == 'admin' and password == 'admin'
endHPC integration
Given SequenceServer simply runs NCBI BLAST+ commands in the shell it's relatively easy to devise a scheme to run BLAST searches on another, more powerful computer or on cluster. For example, by replacing BLAST+ binaries with a "shim" like below, we can run BLAST searches on another computer using SSH.
#!/usr/bin/env sh
blast=`basename $0`
param=`echo "$@" | sed "s/\-db\ /\-db\ \'/" | sed "s/\ \-query\ /\'\ \-query\ /"`
ssh hostname /usr/local/bin/$blast $param
Additionally, TMPDIR environment variable must be set to a
directory that's shared between both the machines, e.g., via SSHFS.
Using a job queuing system such as qsub may be a bit
involved depending on the flexibility afforded by the system.
Fortunately, we have a solution for qsub thanks
to Andy Foster
and Loraine Brillet-Guéguen .
Create the following script:
#!/usr/bin/env sh
jobid=`mktemp bl.XXXX`
rm $jobid
rfile=$1
efile=$2
blast=$3
shift 3
param=`echo "$@" | sed "s/\-db\ /\-db\ \'/" | sed "s/\ \-query\ /\'\ \-query\ /" | sed "s/\-outfmt\ /\-outfmt\ \'/" | sed "s/\ \-num_threads\ /\'\ \-num_threads\ /"`
qsub -sync y -b y -pe slowpara 4 -N $jobid -o $rfile -e $efile /usr/local/bin/$blast $param
And add the following at line 51 of lib/sequenceserver/blast/job.rb:
def run
system("/path/to/script #{stdout} #{stderr} #{command}")
end
As above, TMPDIR environment variable must be set to a
directory that's shared between both the machines, e.g., via a shared
file system such as GPFS, NFS mount or SSHFS.
Embedding SequenceServer in an iframe
By default, any website can embed your SequenceServer installation via iframe provided there is a public IP or URL pointing to it.
You can change this behaviour by setting :frame_options:
key in the config file:
- :frame_options: :deny
- Completely disable embedding in an iframe at all.
- :frame_options: :sameorigin
- Only allow websites hosted within the same domain to embed SequenceServer.
- :frame_options: 'ALLOW-FROM my-url'
- Only allow the webiste hosted at 'my-url' to embed SequenceServer. Of course, 'my-url' is a website address that you provide.
Using SequenceServer's API
SequenceServer has a simple API that you can use to run BLAST searches programatically. Thanks to Richard Adams, the API is documented at the following link, including an example bash script to BLAST all databases: SequenceServer API .
Debugging SequenceServer
If you are making custom modifications to SequenceServer, following tips may come handy:
SequenceServer's development mode, activated as sequenceserver
-D enables verbose logging and loads unbuilt assets (JS and
CSS). SequenceServer's interactive command-line mode, activated as
sequenceserver -i lets you access all server-side objects
and methods, call them and inspect their output in Ruby.
Where is job data stored?
SequenceServer stores job data in ~/.sequenceserver
folder. Each job gets its own directory here and has a
UUID
for name, which is also the job id that is used internally to look up
job status, etc.
Known issues and limitations
- View sequence link is disabled if the length of the hit exceeds 10,000 residues - ok if target sequences are proteins or contigs. We feel this mode of visualising sequences is not optimal for very long sequences (e.g., scaffolds).
- During setup on some versions of OS X, an extra space is added at the end of autocompleted paths when SequenceServer prompts for paths to the BLAST+ executables or database directory. This appears to be due to a bug in Ruby readline library. Unfortunately it is beyond our scope to fix this slightly inconvenient bug, especially since working around it is straightforward (i.e. you just need to backspace it).
Other frequently asked questions (FAQ)
- 0. What about the SequenceServer Cloud Service?
- There are many more FAQs, and dedicated premium support channels for the Cloud BLAST Service!
- 1. Can I use SequenceServer as an access-point for a community genome database?
- Yes. SequenceServer is used as data querying mechnism in over 30 community databases . You can use SequenceServer as it is along with supporting pages describing the data and related resources (e.g., HopBase), customise i t extensively (e.g., Lotus Base) , or integrate it with InterMine (e.g., PlanMine) .
- 2. Does SequenceServer include a genome browser?
- No, but any web based genome browser such as JBrowse , Biodalliance, o r igv.js can b e used. Also see: Integrating with JBrowse and Adding links to search hits .
- 3. Is it possible to disable Grammarly for the query sequences?
- Yes, but each user would have to do it themselves: disable Grammarly .
Understanding BLAST
BLAST is a heuristic, i.e., it is fast and approximate instead of being slow and perfect. It starts by looking for a minimal 100% match (e.g., 11 consecutive nucleotides with 100% identity between your query and the database sequence). If it finds none its over. If it does find a match, it extends that in both directions: identical (or similar) bases add points; differences are negative points. If too many points are lost, it stops aligning. BLAST might not stop at the exact best place, alignment ends might be wrong. bitscore is the total number of points for the aligning region. The bigger it is, the stronger the alignment. But the bitscore doesn't take into account sequence length nor database size. The E-value does take these into account. It is better to look at E-values than bitscores. The E-value represents the number of times the observed alignment would be expected to occur by chance (it is not a p-value!); depends on the bitscore, the length of the query sequence, and the cumulative length of all sequences in the database. It is easier to talk about strong E-values (e.g. 1e-100 = 10-100 = almost zero; impossible to obtain by chance) vs weak E-values (e.g 0.1; for similarity that may be due to chance) than small vs large (which is always a bit confusing).
BLAST has been rewritten several times - most recently by NCBI as BLAST+. NCBI now use and recommend using BLAST+. The BLAST+ publication explains why BLAST+ is easier to use and faster than the old legacy BLAST. WU-BLAST is now commercial and called AB-BLAST. There is probably no good reason to use either alternative. Note that the output formats change slightly from one BLAST implementation to the next. NCBI's BLAST+ is actively developed and is the only one supported by SequenceServer.