Generating guide RNAs and sequencing primers for CRISPR knockouts using BLAST and SequenceServer
Why create animal knockouts?
Gene knockouts enable geneticists to understand the role of a gene in a normal physiological setting as well as the consequences of its truncation or complete loss. Knocking out genes in animal systems, for example, can help us understand how the proteins they encode interact to shape development, behavioral traits, or contribute to disease.
The CRISPR-Cas approach for targeting and knocking out a specific genomic location
The CRISPR-Cas system has completely changed how genome editing is done. This gene editing system includes:
- The Cas enzyme that cuts DNA. It’s a sequence‐specific endonuclease enzyme that introduces double‐strand breaks into DNA.
- Guide RNAs (gRNAs). These gRNAs are the system’s GPS: they ensure the correct genome location is targeted. One single gRNA (sgRNA) or multiple gRNAs targeting or flanking the targeted sequence are used. An sgRNA is a synthetic RNA molecule that combines the functions of what previously required two RNAs: a CRISPR RNA (crRNA) and a trans-activating CRISPR RNA (tracrRNA). Using sgRNA combines these into a single molecule, simplifying the delivery of CRISPR components into cells for gene editing (Jinek M et al., 2012; Motoche-Monar C et al., 2023).
Injecting Cas and the sgRNA creates knockouts because it results in the deletion of a few nucleotides of DNA in the targeted location.
To summarize the process after you’ve designed the sgRNA, you:
- Inject Cas enzyme and sgRNA.
- The Cas enzyme forms a complex with the sgRNA.
- The sgRNA (with attached Cas) binds to the targeted DNA sequence.
- The Cas enzyme looks for a nearby “protospacer-adjacent motif” (PAM) sequence in the DNA and cuts both DNA strands. It can cut twice i.e. double-stranded breaks, leaving blunt ends, or potentially an overhang of up to 3 nucleotides. When Cas cuts twice, some of the DNA may be deleted from the genome.
The following schematic illustrates the CRISPR process (modified from AddGene):

To create a knockout, you typically target a gene’s coding sequence. Double-stranded breaks in DNA can result in a frameshift, where a deletion of one or two base pairs disrupts the triplet reading frame of coding sequences. This can lead to nonsensical proteins that do not function in the same manner as unmodified proteins. Alternatively, frameshifts can induce early stop codons, which results in an early termination of the gene and a truncation of the encoded protein. Whole genomic regions can also be removed by using two different sgRNAs that cause two double-stranded breaks that are repaired via non-homologous end joining.
Knocking out a gene using the CRISPR-Cas system
1. Choosing which Cas endonuclease to use.
Cas9 and Cas12a are the main Cas enzymes used for genome editing. They have different activities, which are important to consider depending on type of DNA break, target gene GC-bias and delivery preference. There are three main differences between these Cas9 and Cas12a (Swarts DC et al., 2018).
- A) Type of DNA break: Cas9 will generate a blunt DNA double-strand break: both strands of DNA are cut in the exact same place. In contrast, Cas12a generates staggered breaks, also known as sticky ends.
- B) DNA cut site: The cutting location is always a few nucleotides from the target sgRNA, at a DNA sequence called a protospacer-adjacent motif element (PAM element). PAM elements are short and conserved sequences of 2–5 base pairs. Cas9 and Cas12a cut at different PAM elements:
- i. Cas9 cuts a PAM sequence of
NGG, where “N” means “any nucleotide” - ii. Cas12a cuts
TTTV, where “V” meansA,CorG
- i. Cas9 cuts a PAM sequence of
- C) Guide RNA requirements: the requirements of gRNA differ between Cas9 and Cas12a enzymes. Cas9 requires a sgRNA complementary to the target DNA, including a PAM sequence for target recognition. In contrast, Cas12a requires a crRNA array which is processed into mature crRNA and does not require an additional tracrRNA or tracrRNA component (Zetsche B et al., 2015)
Many mammalian coding sequences are GC-rich, which makes it easier to find the requisite NGG PAM sequence for guiding Cas9 to the desired location. Bacterial genes typically have higher AT-content, often making it easier to find Cas12a’s TTTV PAM sequence.
2. Designing the sgRNA
To direct the Cas enzyme to the target site in the genomic DNA, the sgRNA must be optimally designed. However, sgRNA design depends on which Cas enzyme is used.
- Cas12 is a single RNA-guided endonuclease, which means it processes its own guide RNAs. It requires a single targeting crRNA that is complementary to one strand of the target DNA sequence.
- Cas9 is more complex - it historically required using two gRNAs: a crRNA and a tracrRNA, essential for the Cas9 system’s assembly. But the system has been simplified, so most people combined the two gRNAs into a single “sgRNA” molecule.
- The sgRNAs need to contain the reverse complementary sequence to the target region of the genome. The target region must also have an adjacent PAM element sequence.
3. Testing for possible off-target binding of sgRNAs with SequenceServer
Once we have designed sgRNAs for a particular genomic region we need to check for off-target binding. Off-target bindings are other genomic regions that exhibit a similar or the same DNA sequence as our designated target site. This is a big issue, as we do not want to additionally edit other parts of the genome. Doing so could disrupt the organisms biology and lead us to draw incorrect conclusions about the impacts of our target.
To assess off-target binding we can take an approach similar to that used for ensuring specificity of PCR primers. We can BLAST our sgRNA sequence against a genome. For the highest sensitivity and specificity, use the parameters that are most appropriate for searching short oligonucleotide sequences, including primers. In SequenceServer we just select the “short oligonucleotides” option from the “Advanced Parameters” drop-down menu.
Using this approach, we can find sites in the genome with high sequence similarity to our target region that might lead to off-target edits.

4. Synthesizing of sgRNAs
The CRISPR-Cas system requires the synthesis of a large amount of sgRNA. To achieve this, specific promoters are used to enable transcription, and depend on whether synthesis is performed in vitro or in vivo. An example of an in vitro construct for sgRNA synthesis is a linear double-stranded DNA molecule. It contains a transcriptional promoter (5’ end), followed by the target sequence, and a universal oligo binding site (3’ end).
5’ Promoter sequence - TARGET SEQUENCE - Universal oligo binding site 3’
Promoter: There are many different promoters, with the T7 promoter most frequently used for driving transcription of target genes or RNA molecules. T7 is a well-characterised and efficient promoter used in molecular biology and biotechnology.
Universal Oligo binding site: The universal oligo binding site at the 3’ end is a short sequence that facilitates the hybridisation of the sgRNA with the Cas9 protein. Tools such as CRISPRscan may be used to help generate the final completed sgRNA sequence. Once this construct is made, large amounts of sgRNA can be synthesized and used for the delivery of CRISPR-Cas.
5. Delivering the Cas enzyme and sgRNA
Cas9 and the sgRNAs can be incubated together in order to form a ribonucleoprotein complex (RNP). This approach reduces off-target effects compared to delivering the same components without prior incubation.
Successful delivery of the CRISPR-Cas system and transformation of the genome of a one-cell embryo, following fertilisation, leads to all somatic and germline cells in the resulting organism to have the genetic change. However, if transfection occurs at a later stage, it is likely that only some cells will be transformed. This organism ends up being a mosaic - with some wild-type cells, and some that have been edited.
Depending on the research question and organism, the RNP complex is delivered by microinjection, electroporation, or lipofection.
- Microinjection uses a sharp needle to perforate a fertilised cell at the one-cell stage in order to introduce the RNP and has been used in generating various knockout animal models, such as zebrafish.
- Electroporation generates pores in the cell membrane to allow for the entry of the RNP into the cytoplasm; this has been used in cell culture and genome editing of mouse zygotes.
- Lipofection is still being developed. This technique involves the use of liposomes or lipid-based nanoparticles to encapsulate nucleic acids and facilitate their entry into the cell membrane.
6. Using SequenceServer to design optimal PCR primers to detect CRISPR edits.
To detect the edits made by CRISPR-Cas, called crispants, we need to sequence the target region. To do this we can use PCR amplification of the target region, followed by sequencing. Once we have designed the PCR primers it is important to check for non-specific amplification and mis-priming. This can be checked in SequenceServer by BLASTing your primers to the genome of your chosen organism. If your target region is the only good hit, and there are no other hits, or only weak hits, then you may have designed good primers.
Example: Knocking out the mab21L2 eye-development gene in zebrafish.

As an example, we’re interested in the MAB21L2 gene, which is required for normal eye development in humans. Let’s say that in humans, an allele exists that leads to an early stop codon in this gene, and thus a highly truncated protein. That mutation changes a “G” at position 339 in the coding sequence to an “A” (c.339G>A). This changes the encoded amino-acid from a Lysine to a STOP codon (p.Trp113Ter), leading to a truncated protein and a loss of function.

Detecting human-zebrafish MAB21L2 orthologs with SequenceServer
We want to develop a zebrafish model for this mutation, in order to better understand its impact. Since we want to make a double-stranded break to induce STOP codons through frameshifts, the Cas9 endonuclease would be suitable. However, we first need to find the zebrafish ortholog of the human MAB21L2 gene. SequenceServer enables us to create a custom BLAST database using the zebrafish genome assembly, and to identify and retrieve our gene of interest from this assembly.
First, we must identify the corresponding gene and mutation in zebrafish:
- We BLAST the gene of interest against the genome or coding sequences of all zebrafish genes, and recover the FASTA sequence of the zebrafish gene.
- Then, find the location of interest where the mutation is in the FASTA input to locate it in the zebrafish genome.
When we BLASTN the human gene’s coding sequence against the zebrafish genome, there is a strong top hit on chromosome 1, followed by a weaker hit on chromosome 15. For the top hit, a combination of small E-value, high query coverage, total score and % identity is a good indicator of 1-to-1 orthology. Conversely, the hit at chromosome 15 which only has 79% identity (instead of almost 100%) is likely to be a paralog.

One way of checking orthology relationships is to do a reciprocal BLAST. For this, we retrieve the top hit from the zebrafish genome. We can easily download the zebrafish hits from the left-hand dropdown menu in SequenceServer, and use them to BLASTN them against the human genome.

The reciprocal BLAST shows that the human MAB21L2 gene and the zebrafish mab21L2 gene are both the top hit in their respective genomes. Being “top reciprocal BLAST hits” is a good sign of orthology. If the pattern were more complex, we would need to consider performing a phylogenetic analysis encompassing multiple putative homologous genes from several species to be more confident.
Determining an optimal sgRNA target with SequenceServer
Now that we have the human-zebrafish orthologs of MAB21L2, we need to determine where to make the edits in the zebrafish genome. The SequenceServer BLAST screenshot below shows how the human (Query) sequence aligns to the zebrafish genome (Subject). The codon involving the c.339G>A mutation in humans is circled in red.
Fortunately, there is a PAM domain closeby, and is where Cas9 will make the cut. This NGG PAM domain, here TGG, is circled in blue. We can then design our sgRNA to target the sequence upstream, circled in green.
Once the sgRNA has been designed, the most likely resulting frameshift variants can be assessed using online tools, such as inDelphi, to assess the probability of a successful frameshift using the sgRNA designed and its efficacy to cause a truncated protein.

Producing the MAB21L2 sgRNA
To transcribe the sgRNA we can use the T7 promoter. This is placed 5’ upstream of the sgRNA sequence. On the 3’ a universal oligo binding site is also added. The complete sgRNA structure and sequence follows:
5’ T7 promoter sequence - TARGET SEQUENCE - Universal oligo binding site 3’
5’ taatacgactcactataGAGGAGCATGTCCCTGTGGGgttttagagctagaa 3’
Testing for possible off-target binding of our sgRNA with SequenceServer
Our BLAST analysis above indicated that there is a paralogous gene with relatively high similarity to our target gene. This increases the risk of our sgRNA binding off-target.
We can take an approach similar to that used for ensuring specificity of PCR primers: Let’s BLAST our sgRNA sequence against the zebrafish genome. For the highest sensitivity and specificity, use the parameters that are most appropriate for searching short oligonucleotide sequences, including primers. In SequenceServer we just select the “short oligonucleotides” option from the “Advanced Parameters” drop-down menu.
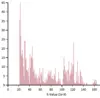
It initially looks like we have many hits. The figure below illustrates multiple hits with varying Total and E-value scores. Notably, the genomic location of interest on chromosome 1 exhibits a significantly stronger (i.e., lower) E-value of 5.54x10-3 compared to the next closest hit on chromosome 3, which has a weaker (i.e., greater) E-value of 5.34. Despite this, both hits display large total scores. Other off-target sites identified also show high total scores and large E-values. The weak E-values of these hits indicate that they are spurious, rather than representing a genuine biological relationship. If we examine the pairwise alignments, we see that the aligning segments are only a small portion of our query sequence. High similarity over a small segment will mean much lower binding affinity than complete similarity over the entire query sequence. In particular, because the alignment doesn’t include the last nucleotides of our sgRNA, it is unlikely that we would get much non-specific binding to these additional sites.

For easy interpretation, SequenceServer provides graphic representation of pairwise alignments of query and hit sequences, showing how each aligning segment (High Scoring Pair - HSP) aligns to the query. Below are some example alignments from the top two hits on chromosome 1 and 3, highlighting the differences in E-value’s significance between the other genomic locations compared to the location of interest, highlighted in red.

The graphical overview of aligning regions shows how the query (top) aligns to different genomic regions of the zebrafish genome (bottom), with darker segments representing stronger similarity. In this example, the stronger query-subject hit is located on chromosome 1 around position 24 Mbp. The pairwise-alignment of this hit (highlighted in red) further confirms the high similarity between query and subject.
Less significant hits (i.e., off target sites) can be easily identified thanks to SequenceServer’s graphical overview.

Using SequenceServer to design optimal PCR primers to detect CRISPR edits.
After we’ve transformed our fish, and thus created “crispants” (i.e., from CRISPR-Cas mutants), we typically want to check which exact changes occurred. For this, we must Sanger sequence the target region. To do this we can use PCR amplification of the target region, followed by sequencing. Once we have designed the PCR primers it is important to check for non-specific amplification and mis-priming. This can be checked in SequenceServer by BLASTing your primers to the genome of your chosen organism. If your target region is the only good hit, and there are no other hits, or only weak hits, then you may have designed good primers.
Conclusion
Overall, knockout animal models are indispensable tools for advancing our understanding of gene function, disease mechanisms, and therapeutic interventions in biological and medical research. They provide valuable insights into the complex interactions between genes, development, and disease, ultimately contributing to the development of new treatments and therapies. In order to harness these biological and pharmacological insights, SequenceServer can be used to help create specific and effective sgRNAs for gene knockdowns for phenotypic and functional analyses.
Happy BLASTing!
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., & Charpentier, E. (2012). ‘A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity’, Science, 337(6096), 816–821. doi: 10.1126/science.1225829.
Motoche-Monar, C., Ordoñez, J. E., Chang, O., & Gonzales-Zubiate, F. A. (2023). ‘gRNA Design: How Its Evolution Impacted on CRISPR/Cas9 Systems Refinement’, Biomolecules, 13(12). doi: 10.3390/biom13121698.
Swarts, D. C., & Jinek, M. (2018). ‘Cas9 versus Cas12a/Cpf1: Structure-function comparisons and implications for genome editing’, Wiley interdisciplinary reviews. RNA, 9(5), e1481. doi: 10.1002/wrna.1481.
Zetsche, B., Gootenberg, J. S., Abudayyeh, O. O., Slaymaker, I. M., Makarova, K. S., Essletzbichler, P., Volz, S. E., Joung, J., van der Oost, J., Regev, A., Koonin, E. V., & Zhang, F. (2015). ‘Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system’, Cell, 163(3), 759–771. doi: 10.1016/j.cell.2015.09.038.